Research Interests
- Pathogenic mechanisms underlying epilepsy mutations in Kv7 channels
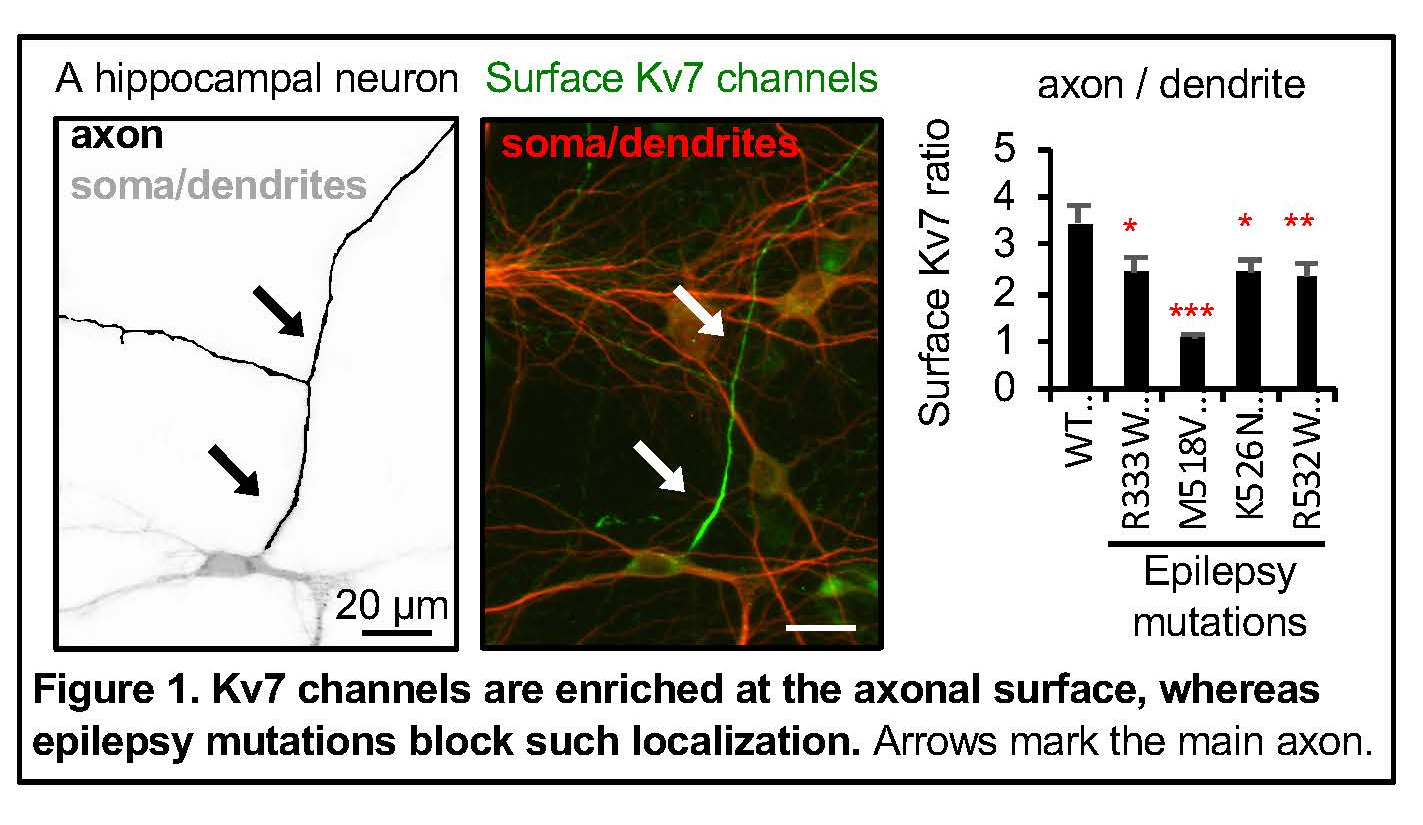
Neuronal Kv7/KCNQ potassium channels are heterotetramers composed of Kv7.2/KCNQ2 and Kv7.3/KCNQ3 subunits, which are enriched in the hippocampus and neocortex, the brain regions important for learning and memory. Subcellularly in neurons, they are preferentially enriched at the axonal plasma membrane compared to somatodenritic surface with highest concentration at the axonal initial segments where action potential initiate. These channels give rise to slowly activating and non-inactivating voltage-dependent outward potassium currents (M-current) which potently inhibit repetitive and burst firing of action potentials and regulate resting membrane potentials.
Physiological significance of these channels are underscored by the fact that dominant mutations in KCNQ2 and KCNQ3 genes cause early-onset epilepsy including benign familial neonatal epilepsy (BFNE) and epileptic encephalopathy (EE). In contrast to transient appearance of seizures in neonates with BFNE, EE is characterized by seizures that are often drug-resistant, psychomotor retardation, developmental delay, intellectual disability, autism, speech delay, and neuroradiological abnormalities including white matter reduction and enlarged ventricles. Since the mapping of the first BFNE variants to Kcnq2 gene in 1998, > 300 mutations in KCNQ2 and KCNQ3 genes have been associated with epilepsy. However, how these epilepsy mutations ultimately lead to epilepsy with variable clinical severity remain largely elusive.
One major research goal in the Chung lab is to determine the pathologic mechanisms underlying epilepsy mutations of KCNQ2 and KCNQ3.
The major questions are:
- Do epilepsy mutations affect voltage-dependent activation and axonal targeting of Kv7 channels and disrupt their ability to inhibit neuronal excitability?
- How does the disruption of Kv7 channel function and expression ultimately lead to epilepsy in mice?
- Why certain mutations result in BFNE but other mutations lead to EE?
 To investigate, we employ interdisciplinary methods including structure-function studies, imaging, biochemistry, electrophysiology in neuronal culture and transgenic mice.
To investigate, we employ interdisciplinary methods including structure-function studies, imaging, biochemistry, electrophysiology in neuronal culture and transgenic mice.
The Chung Lab discovered that calmodulin binding to helices A and B of Kv7.2 mediates Kv7 targeting from the endoplasmic reticulum (ER) to the axonal surface (Cavaretta et al., 2014). We developed novel statistical algorithm to identify mutation hotspots for epileptic encephalopathy (EE) in Kv7.2, and showed that select EE variants in these hotspots impaired the function of its associated protein domain including voltage-and PIP2-dependent gating and axonal trafficking (Kim et al., 2018; Zhang et al., 2020). One EE mutation in calmodulin binding domain in helix B of Kv7.2 induced degeneration of hippocampal neurons in culture (Kim et al., 2018), resulting in spontaneous seizures and cognitive deficits (Kim et al., PNAS, 2021). We also showed that heterozygous loss of Kv7.2 induces abnormal behaviors reminiscent of autism (Kim et al., 2019).
- Identification of molecular mechanisms underlying homeostatic intrinsic plasticity
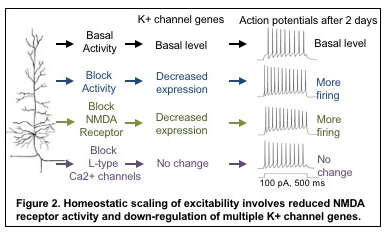
Electrical properties of neuronal membranes can undergo persistent modification in response to neuronal activity or sensory experience. Homeostatic plasticity is one such mechanism by which neurons adapt their electrical activity within a physiologic range based on their previous activity. For example, a 2-day blockade of neuronal activity in the hippocampus leads to compensatory increase in neuronal communication at synapses (termed synaptic transmission) as well as the ability of hippocampal neurons to fire action potentials at a given input signal (termed intrinsic excitability). Interestingly, activity blockade in the hippocampus for 2-4 weeks leads to temporal lobe epilepsy in rodents. These findings raise a compelling question: “when and how do neurons exploit homeostatic plasticity to stabilize their network as a normal adaptive response, or to cause persistent neuronal hyperexcitability as a pathological manifestation in epilepsy?”
One major research goal in the Chung lab is to answer this question by first determining how homeostatic plasticity is induced in the normal brain.
The major questions are:
- What ion channels are regulated during the induction of homeostatic plasticity ?
- What are the signaling pathways that sense changes in neuronal activity level and convey to the homeostatic changes in synaptic transmission and intrinsic excitability?
- How does homeostatic regulation in ion channels (or their dysregulation) contribute to acquired epilepsy?
To investigate this, we employ interdisciplinary methods including imaging, biochemistry, electrophysiology in neuronal culture and transgenic mice in which activity blockade can be induced genetically.
We discovered that induction of homeostatic scaling of intrinsic excitability induces current and transcript reduction of multiple K+ channels including Kv1 and Kv7 and involves signaling pathways distinct from homeostatic synaptic scaling (Lee et al., 2014; Lee et al., 2015). We were the first to identify genes regulated during homeostatic plasticity (Lee et al., 2015).
One of the genes was PTPN5, which encodes brain-specific tyrosine phosphatase STEP61. We discovered that activity-dependent downregulation of STEP61 and up-regulation of NMDA receptor subunit GluN2B and AMPA receptor subunit GluA2 contributes to homeostatic scaling of excitatory synaptic strength (Jang et al., 2015). In contrast, prolonged enhancement of neuronal activity in culture and electroconvulsive seizures increase STEP61 and decrease tyrosine phosphorylation of GluN2B and GluA2 (Jang et al., 2015; Jang et al., 2016).
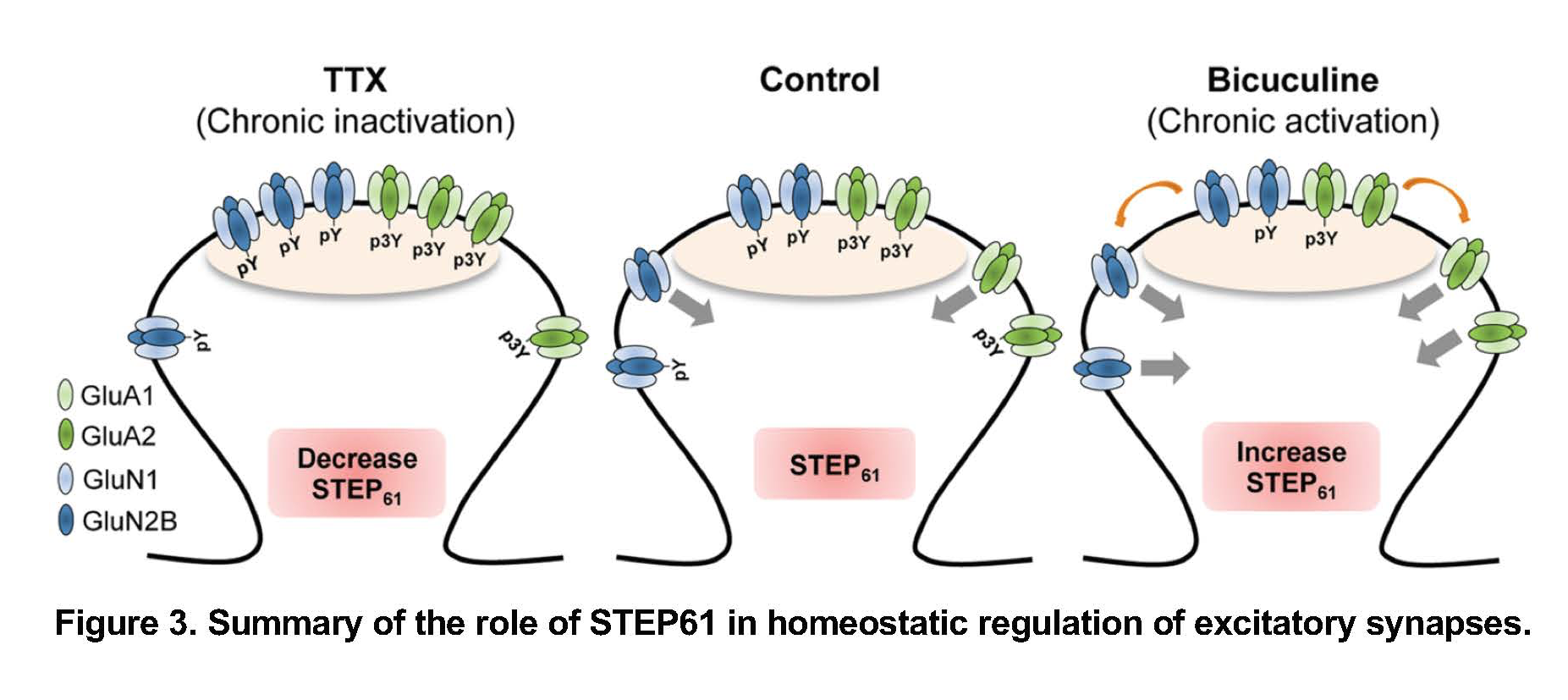
Currently, we are testing if STEP61 controls intrinsic excitability, and susceptibility to seizures in mouse models of TBI and epilepsy.
- Identification of novel molecular mechanism in Alzheimer’s disease.
The Chung lab is interested in delineating the mechanisms underlying the hyperexcitability and synaptic dysfunction in early stage of Alzheimer’s disease (AD) due to their strong association with memory loss before neurodegeneration is evident.
The Chung lab launched a new research in AD in collaboration with the Selvin and Kong labs in Physics and Chemical Engineering to (1) use super-resolution imaging to understand soluble Aβ and hyperphosphorylated tau affect glutamate receptors, STEP61, and KCNQ/Kv7 potassium channels in the presence of Apolipoprotein-ԑ4 (ApoE4), the most prevalent genetic risk factor for AD, and (2) test Dr. Kong’s novel nanoparticles as drug carriers to correct ApoE4 and alleviate synaptic dysfunction and neuroinflammation in AD.